
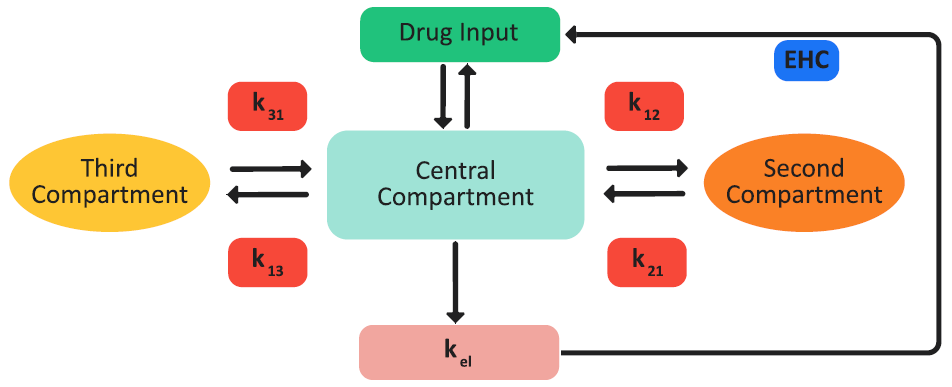
If the necessary pharmacokinetics (PK) parameters are included in the project, then the GastroPlus® model can carry out one-, two-, or three-compartment pharmacokinetic simulations of the drug concentration in plasma, Cp, versus time. The pharmacokinetic model can account for plasma protein binding (fup%), blood to plasma drug concentration ratio (Rb:p), fixed first pass extraction (FFPE%) for both the intestine and liver, and renal clearance (CLrenal). Elimination from the central compartment in any of these models can follow either linear or non-linear (saturable) kinetics in any of these models. Figure 2-59 is a schematic of a three-compartment pharmacokinetics in the GastroPlus® model.
Figure 2-59: Three-compartment pharmacokinetic GastroPlus® model
-
One-compartment pharmacokinetic model: The drug is assumed to be distributed in a single pharmacokinetic compartment.
-
Two-compartment pharmacokinetic model: The drug can be distributed in an additional compartment. The transfer between the two compartments is modeled by a first-order equation and is parameterized based on the distribution rate constants, k12 and k21. The elimination of the drug occurs only from the central compartment.
-
Three-compartment pharmacokinetic model: A third compartment that is similar to the second compartment is also available for the distribution of the drug from the central compartment. The second and third compartments do not interact. The elimination of the drug occurs only from the central compartment.
Traditionally, absorption and systemic disposition have been studied separately to describe in vivo drug behavior. Although this assumption can clearly be used for many drugs, this is not the holistic reality. The concentration gradient-driven absorption in the ACAT model clearly shows that absorption and systemic disposition interact. The correct understanding and prediction of drug pharmacokinetics requires a model that solves both absorption and systemic distribution simultaneously.
Enterohepatic Circulation (EHC) with compartmental PK models
Enterohepatic Circulation (EHC) can be modeled as part of the pharmacokinetics of any drug. With Compartmental PK, the GastroPlus® EHC model (Figure 2-60) diverts a fraction of the drug that is cleared from the central compartment to a loop that includes a gallbladder for release after a meal. This diverted amount is calculated as the total systemic clearance multiplied by a fitted number called the biliary clearance fraction (fbcl).

Because a rat does not have a gallbladder, the circulation for rat is modeled as continuous in the GastroPlus® EHC model.
For species with a gall bladder, the diverted fraction of drug is split into two parts. One part is directed to the gallbladder where it is stored during the fasted state. This part is calculated as the biliary clearance fraction (fbcl) multiplied by a gallbladder fraction (fgb). The remaining part continuously recirculates to the duodenum, where it is available for re-absorption.
Figure 2-60: EHC model in GastroPlus®
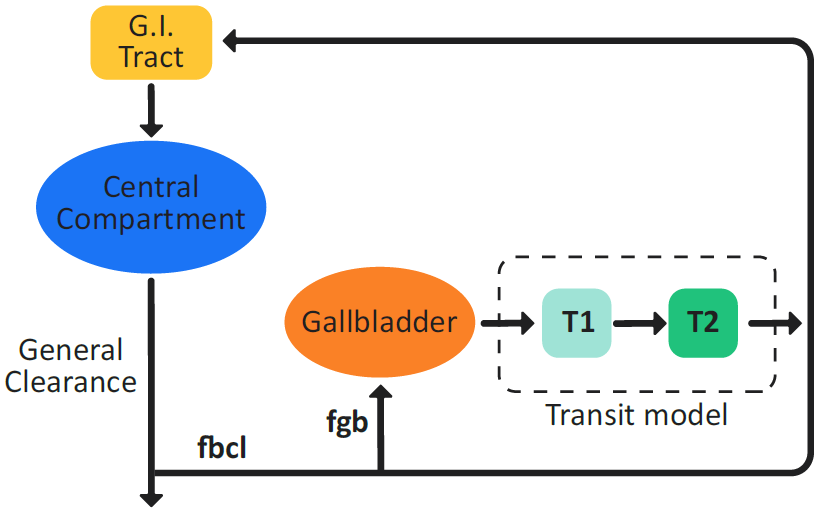
A switch from a fasted state physiology to a fed state physiology triggers gallbladder emptying. The gallbladder empties its contents into the duodenum using a Gaussian release-time model to simulate gallbladder contraction, which is driven by the Gallbladder Emptying Time (GET).
In addition to the EHC that is induced by a meal, a specified fraction of the amount of drug that is going into the EHC loop continuously enters the duodenum, even in the fasted state. This fraction is the complement of the gallbladder diversion fraction (fgb).
For a discussion of EHC implementation with PBPK models, see Liver tissue including enterohepatic circulation.