
The Simulations view is where all previously created assets are brought together to create a simulation. This view has four panels. The first panel, Drug Administration, contains settings for which Compounds, Dose Schedules, and Physiology Schedules will be used. The Compound Settings panel provides options for controlling the simulation settings to be used for each compound identified in the Drug Administration panel. The Additional Dosage Routes panel provides options specific to each individual additional dosage route. Lastly, the Configuration panel allows users to set the simulation length and other computational settings for running the simulation.
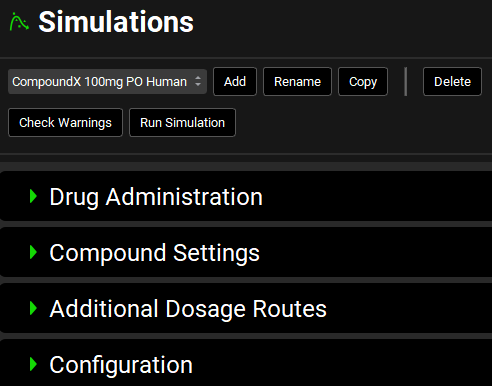
Simulations view
|
Input/Option |
Description |
|
Simulation Name |
A drop-down. Selects the current simulation. Options are populated based on simulations already added to the project. If no simulations have been added, this will be blank, and panels below will be disabled.
|
|
Check Warnings |
Checks the simulation setup and displays any warnings in the message center. Warnings may be critical, for example trying to run a PBPK simulation without a PEAR physiology set up or trying to use a compound-physiology pair for which pharmacokinetic properties have not yet been defined, or informational, for example, the simulation time is shorter than that defined in the Dosing Schedule. |
|
Run Simulation |
Runs the current simulation. After the simulation is complete, the interface will automatically navigate to Analysis view, where simulation results will be displayed under the “Exploratory Simulation” run. |
An error message may appear upon creating a simulation for the first time if the compound hasn’t been paired with the physiology yet. (“There are currently no physiolomolecular properties for ‘DrugX’ and ‘PhysiologyX’ ”). Click OK to dismiss the message and navigate to the Pharmacokinetic to pair the molecule to the given physiology.
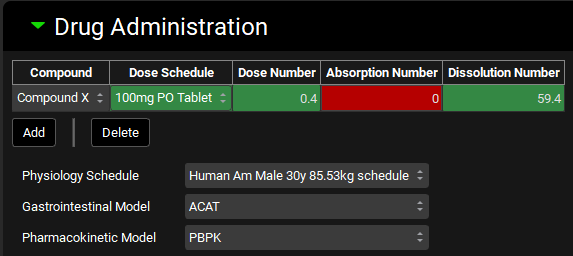
Drug Administration panel
The Drug Administration panel is used to define which Compound(s) will be administered during the simulation, and which Dosing Schedule(s) will be used to administer each compound. Additionally, this is where the Physiology Schedule to use in the simulation is selected.
The desired Gastrointestinal (ACAT or Simplified) and Pharmacokinetic (Compartmental, Simplified, or PBPK) Models to be used in the simulation are also selected here.
The selections made here will affect which options are available in the Compound Settings panel.
Simulations view, Drug Administration panel
|
Input/Option |
Description |
|
Compound |
The compound(s) to be administered in the simulation. Any compound in the project may be added here. Multiple compounds may be administered in one simulation by using the Add button, however interactions between the compounds will not be considered. |
|
Dose Schedule |
The dose schedule which will control how the indicated compound is administered during the simulation. |
|
Dose Number, Absorption Number, and Dissolution Number |
Calculated summary properties based on the combination of the compound, dose schedule, and physiology. Color indicates the estimated favorability for overall drug absorption for oral doses. They will be blank for an IV dose.
|
|
Gastrointestinal Model |
Selects which absorption model to use: ACAT or Simplified. Please note that ACAT applies to both ACAT and ACATPlus models. Specific parameters for each model can be set in the respective Compound Settings sub-panels (See ACAT Model sub-panel and Simplified Absorption sub-panel). If the pharmacokinetic model is set to simplified, only the simplified absorption model may be used. |
|
Pharmacokinetic Model |
Selects which systemic pharmacokinetic model to use: Compartmental, Simplified, or PBPK. Physiologically based simulations can only be used if all PEAR physiologies have been created for all physiologies in the selected Physiology Schedule, and PBPK settings have been setup for each compound-physiology pair. |
|
Physiology Schedule |
Selects which physiology schedule (See Physiology Schedules View) to use in the simulation. |

If pharmacokinetic properties have not yet been defined for the compound-physiology pairs indicated in the Drug Administration panel, the program will prompt for the appropriate pharmacokinetic parameters to be created before running the simulation. This can be done by navigating to the Pharmacokinetics view and selecting the desired compound and physiology at the top of the view.
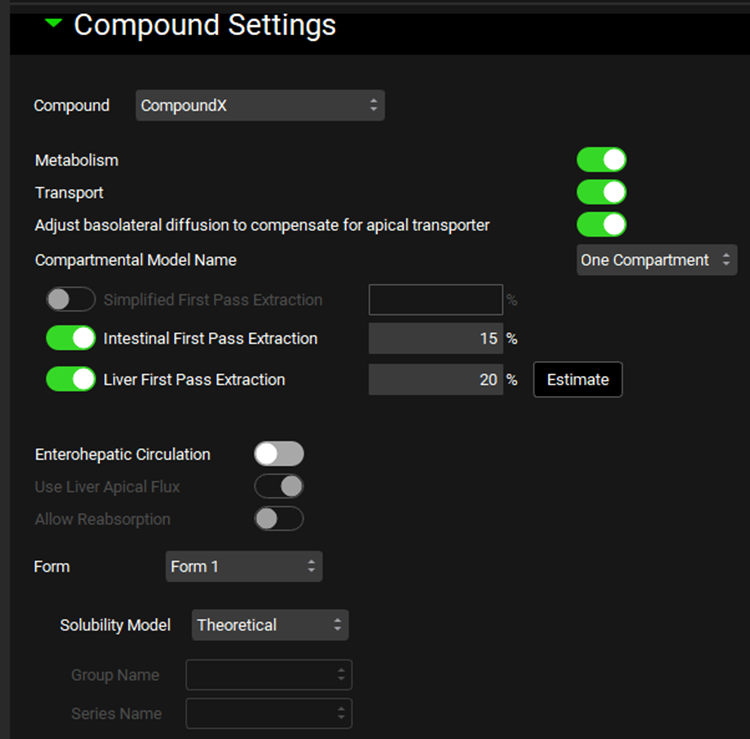
Compound Settings panel
The Compound Settings panel is used to view and set compound-specific simulation settings. These settings control how the various assets available in the project are used in a specific simulation.
Simulations view, Compound Settings panel
|
Input/Option |
Description |
|
Compound |
A drop-down. Selects the compound to which the below simulation settings will be applied. This list is automatically populated from the compounds added to the drug administration table, as well as any metabolites expected to be generated. |
|
Metabolism* |
A toggle. Selects whether all metabolisms (entries in the enzyme kinetics table; see Enzyme Kinetics panel) will be used or ignored for the indicated compound. |
|
Transport* |
A toggle. Selects whether all transports (entries in the transporter kinetics table; see Transporter Kinetics panel) will be used or ignored for the indicated compound. |
|
Adjust Basolateral Diffusion to Compensate for Apical Transporter |
A toggle. Indicates whether basolateral enterocyte flux will compensate for an apical transporter. This allows compounds with low intestinal permeability to exit the basolateral membrane (into the portal vein) if they have apical uptake into enterocytes. On by default. |
|
Compartmental Model Name |
A drop-down. Selects the compartmental model to be used for the compound, based on the models set up in the Pharmacokinetic view->Compartmental panel (See Compartmental Panel). Only enabled for compartmental simulations. |
|
Simplified First Pass Extraction |
Fixed first pass extraction (as a percent of mass) for simplified simulations. Extraction occurs between the simplified dose compartment and the systemic circulation (central) compartment. This value can be edited. |
|
Intestinal First Pass Extraction |
Fixed first pass extraction (as a percent of mass) for compartmental or PBPK simulations using the ACAT model for oral dosage forms. Extraction occurs between the lumen dissolved compartment and the enterocyte compartment. This value can be edited. |
|
Liver First Pass Extraction |
Fixed first pass extraction (as a percent of mass) for compartmental simulations using the ACAT model for oral dosage forms. Extraction occurs between the portal vein compartment and the systemic circulation (central) compartment. This value can be edited.
|
|
Estimate |
Estimates the fixed Liver First Pass Extraction based on clearance and compound and physiological properties. |
|
Enterohepatic Circulation |
A toggle. Controls whether Enterohepatic Circulation (EHC) will be used in the simulation. Off by default. |
|
Use Liver Apical Flux |
A toggle. Controls whether flux across the apical surface of the liver will contribute to EHC in a PBPK simulation. On by default but disabled for non-PBPK simulations. If toggled off, only biliary clearance fraction will contribute to EHC. |
|
Allow Reabsorption |
A toggle. Controls whether compound undergoing EHC will be allowed to transit to the duodenum and reabsorb. If toggled off, transit to the duodenum is disabled and drug will accumulate in the gall bladder only. Off by default. Setting this toggle to off is useful for modeling bile duct cannulation studies. |
|
Form |
A drop-down. Selects the molecular polymorph form of the indicated compound to which the below solubility settings will be applied. Selection also applies to the Nanoparticle Effect and Bile Salt Effect sub-panels below. |
|
Solubility Model |
Selects whether to use the theoretical (Henderson–Hasselbalch based, see Dissociation (pKa) panel) or interpolated solubility model for calculating pH-dependent solubility of the indicated compound and form. If interpolated is selected, the observed data must be specified below. |
|
Group Name and Series Name |
Selects the group and series of observed data to use for the interpolated solubility model. Disabled if theoretical solubility model is selected above. |
*If there is no active license for Metabolism and Transport, these toggles must be OFF in order to run the simulation.
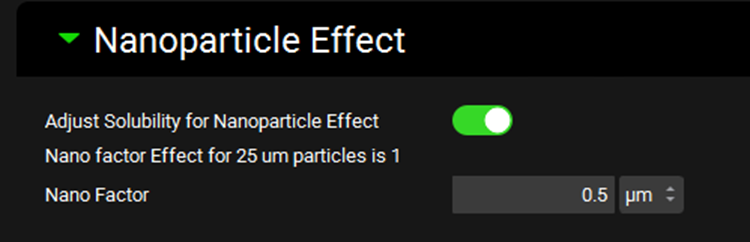
Nanoparticle Effect sub-panel
The Nanoparticle Effect refers to the ability of nano-sized particles to supersaturate in the local environment. This option will enhance the dissolution of formulations with sub-micrometer particle sizes.
Simulations view, Compound Settings panel, Nanoparticle Effect sub-panel
|
Input/Option |
Description |
|
Adjust Solubility for Nanoparticle Effect |
A toggle. Controls whether the nano factor is applied in the dissolution equation to allow for different degrees of supersaturation depending on the particle size. |
|
Nano Factor Effect for [] |
The degree of the nanoparticle effect on the indicated particle size. Particle size is derived from the first formulation in the dosing schedule and is provided here for information only. Where applicable, the nanofactor effect is calculated individually for each particle size during the simulation. |
|
Nano Factor |
A compound-specific empirical parameter, in µm, which is internally scaled to account for particle size. The value can be edited. |
Bile Salt Effect sub-panel
The Bile Salt Effect accounts for the effect of bile salts in vivo on the solubility and diffusion coefficient of lipophilic compounds in the small intestine. The extent of this effect is dependent on both solubilization ratio and bile salt concentration in the relevant gastrointestinal compartment. Solubilization ratio can be determined using one of the three models:
-
Solubility based – calculated based on biorelevant solubilities (See Biorelevant In Vitro Solubilities sub-panel)
-
Mithani equation – estimated based on the LogP of the compound
-
User Defined – entered by the user
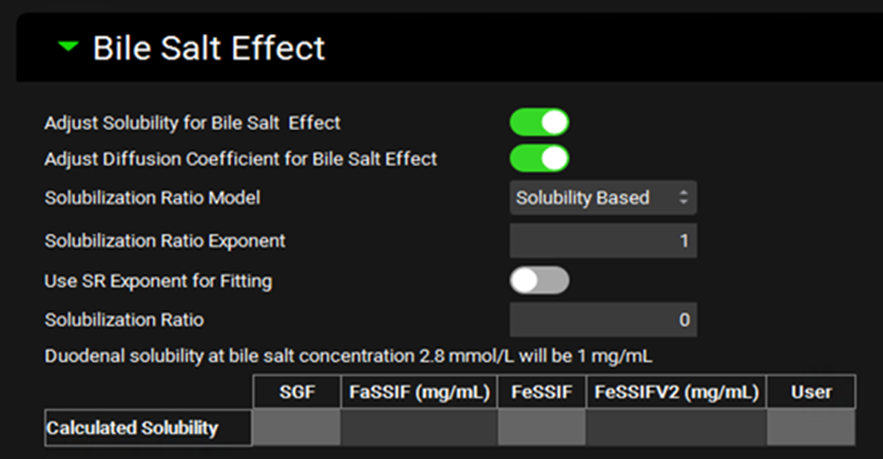
Simulations view, Compound Settings panel, Bile Salt Effect sub-panel
|
Input/Option |
Description |
|
Adjust Solubility for Bile Salt Effect |
A toggle. Controls whether the solubility of the compound will be enhanced in gastrointestinal compartments as a function of bile salt concentration, based on the solubilization ratio indicated below. |
|
Adjust Diffusion Coefficient for Bile Salt Effect |
A toggle. Controls whether the diffusion coefficient, and thus the dissolution, of the compound will be enhanced by the bile salt effect. |
|
Solubilization Ratio Model |
A drop-down. Selects which Solubilization Ratio Model to use: Solubility Based, Mithani Equation, or User Defined |
|
Solubilization Ratio Exponent |
The value of the exponent term that relates the concentration of bile salts to their effect on the solubility of the compound. |
|
Use SR Exponent for Fitting |
A toggle. Controls whether the exponent will be fitted together with the Solubilization Ratio from in vitro measured biorelevant solubilities. |
|
Solubilization Ratio |
The solubilization ratio that will be used in the simulation. For the Solubility Based and Mithani Equation models, this displays the calculated value and cannot be edited. If the model is set to User Defined, then the value can be entered here. |
|
Duodenal Solubility |
Provides the calculated bile salt-dependent duodenal solubility at the indicated bile salt concentration. This is provided for informational purposes only. The solubility will be calculated for each gastrointestinal section during the simulation based on bile salt concentrations. |
|
Calculated Solubilities |
Displays the biorelevant solubilities calculated using the Solubilization Ratio to enable comparison with the measured or predicted values entered on the Biorelevant In Vitro Solubilities sub-panel. |
Dissolution Model sub-panel
The Dissolution Model sub-panel allows users to select which dissolution model to use for the indicated compound in the simulation. The available models are: Johnson, Wang Flanagan, Instant, Fixed Z-Factor, and Interpolated Z-Factor.
The Johnson, Wang Flanagan, and Instant models depend solely on compound properties and do not have further settings. Conversely, the two Z-Factor models require additional settings which can be seen in the figures below.
Simulations view, Compound Settings panel, Dissolution Model sub-panel (with Johnson selected)

|
Input/Option |
Description |
|
Dissolution Model |
A drop-down. Selects which dissolution model to use. The Johnson model is the default selection. Other options are Wang Flanagan, Instant, Fixed Z-Factor and Interpolated Z-Factor. |
The fixed Z-Factor model requires additional input to specify and/or fit the Z-factor. If the Z-factor is already known, all that is needed is to input the value. Otherwise, to fit a Z-factor, one or more In Vitro Dissolution Release profiles must be entered in the Observed Data view (see Observed Data View). These profiles may then be selected for fitting in this sub-panel, where they can be used to fit a single Z-factor of a series of pH-dependent Z-factors.
The series of pH-dependent Z-factors must be fitted within this sub-panel (with Dissolution Model set to Fixed Z-factor and Fit Z-Factor pH Series toggle on) prior to being available for use in the Interpolated Z-factor model
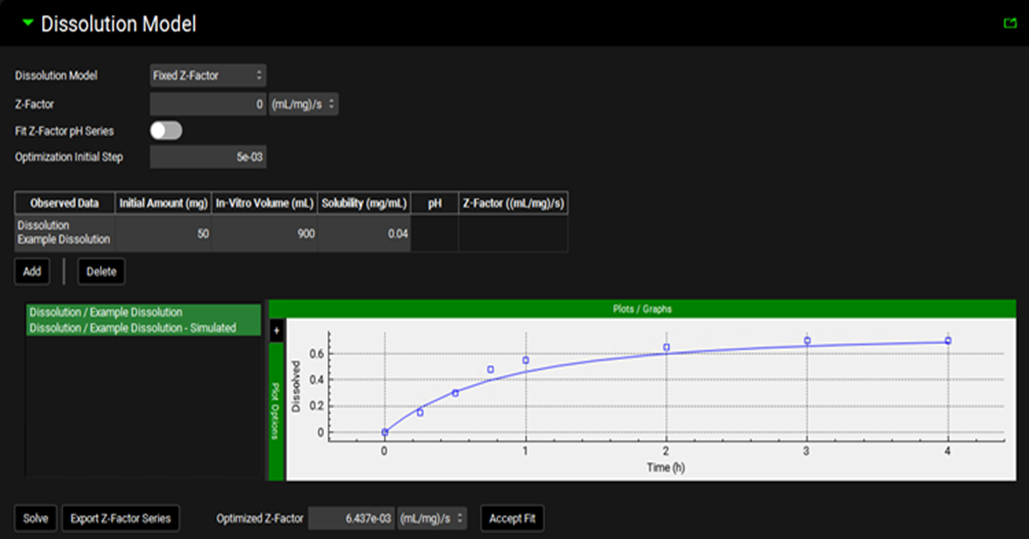
Simulations view, Compound Settings panel, Dissolution Model sub-panel (with Fixed Z-factor selected)
|
Input/Option |
Description |
|
Dissolution Model |
A drop-down. Selects which dissolution model to use. The Johnson model is the default selection. |
|
Z-factor |
The Z-factor, in (mL/mg)/s, which will be used in the simulation. The value can be edited manually, or set via the fitting options below. |
|
Fit Z-Factor pH Series |
A toggle. Selects whether the fitting below should fit a single Z-factor, or a series of pH-dependent Z-factors. Switched off by default indicating that a single Z-factor will be fitted. Switch on to fit a series of pH-dependent Z-factors. |
|
Optimization Initial Step |
The starting step size taken by the optimizer when fitting the Z-factor(s). Default value is 0.005. |
|
Observed Data |
The dissolution time profile(s) from Observed Data to be used in Z-factor fitting, as identified by group and series. Multiple profiles may be used by clicking the Add button. |
|
Initial Amount |
The amount of drug, in mg, initially added to the dissolution experiment. The value can be edited and is required for fitting. |
|
In-Vitro Volume |
The volume of the dissolution medium, in mL, used in the dissolution experiment. The value can be edited and is required for fitting. |
|
Solubility |
The solubility, in mg/mL, of the drug in the media used in the dissolution experiment. The value can be edited and is required for fitting. |
|
pH |
The final pH of the medium in the dissolution experiment. Used only when Fit Z-Factor pH Series is toggled on. The value can be edited and is required when fitting pH-dependent Z-factors. |
|
Z-factor |
Displays the fitted Z-factor for the respective data and pH value. Used only when Fit Z-Factor pH Series is toggled on. Displayed only after solving (see below). |
|
Z-factor Plot |
Plots the observed data series used in Z-factor fitting, as specified in the table above, alongside the predicted dissolution profile using the fitted Z-factor(s) (after fitting). |
|
Solve |
Fits the Z-factor(s) to the data indicated above. |
|
Export Z-factor Series |
Creates a new Dissolution Rate (Z Factor) series in Observed Data (see Profiles Panel) based on the pH-dependent Z-factors fitted here. Relevant only when Fit Z-Factor pH Series is toggled on. The new series can then be used with the Interpolated Z-factor Model. |
|
Optimized Z-factor |
The result of the single Z-factor fitting. |
|
Accept Fit |
Transfers the optimized Z-factor to the Z-factor field above (single Z-factor). |
The interpolated Z-factor model will use a different Z-factor in each gastrointestinal section, based on the pH in that gut section, using Z-factor values interpolated from a Z-factor vs pH profile. Thus, this model requires selection of a Z-factor vs pH profile from Observed Data (see Profiles Panel). The series can either be entered manually in the Observed Data view, or it may fit using the Fit Z-Factor pH Series option under the Fixed Z-factor model.
Simulations view, Compound Settings panel Dissolution Model sub-panel with (Interpolated Z-factor selected)

|
Input/Option |
Description |
|
Dissolution Model |
A drop-down. Selects which dissolution model to use. The Johnson model is the default selection. |
|
Group Name |
The group to which the Z-factor vs pH series belongs. |
|
Series Name |
The name of the Z-factor vs pH series to be used in the simulation. |
Precipitation sub-panel
The Precipitation sub-panel allows users to select which precipitation model to use for the indicated compound in the simulation. Both First Order and Mechanistic models are available, as well as the option to use no precipitation in the simulation. Each model has unique settings.
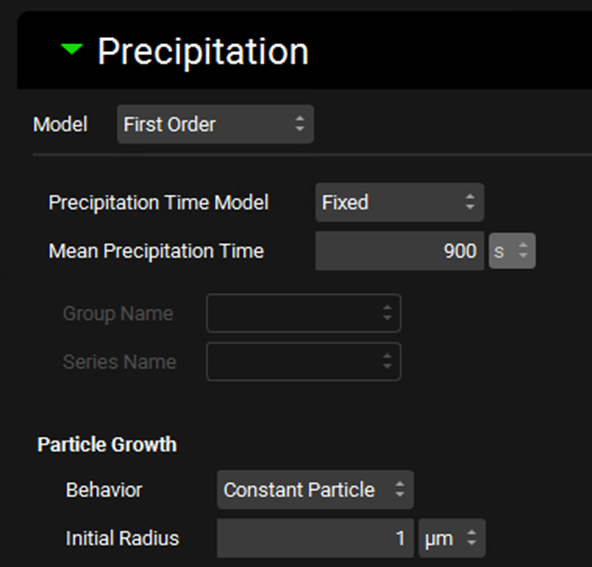
Simulations view, Compound Settings panel Precipitation sub-panel (with First Order selected)
|
Input/Option |
Description |
|
Model |
A drop-down. Selects which precipitation model to use, First Order, None, Mechanistic. First Order is the default selection. |
|
Precipitation Time Model |
Selects whether to use a fixed precipitation time (input as Mean Precipitation Time) or an interpolated model, based on a Precipitation Time vs pH series from Observed Data (see Profiles Panel). If Interpolated is selected the observed data must be selected below. |
|
Mean Precipitation Time |
The mean precipitation time, in seconds, for Fixed Precipitation Time models. The value can be edited. |
|
Group Name and Series Name |
Selects the Precipitation Time data from Observed Data to be used in Interpolated Precipitation Time models. |
|
Particle Growth |
Settings controlling the growth of precipitated drug particles. |
|
Behavior |
A drop-down. Selects the method of particle growth with respect to particle radii. Options are Constant Particle, First Bin (grows particles in first bin only), or All Bins (grows all particles). |
|
Initial Radius |
Sets the initial radius, in µm, of newly formed precipitate particles. The value can be edited. |
The Mechanistic precipitation model requires different parameters to control the mechanistic nucleation and growth model. Two versions are available: Lindfors and Simulations Plus.
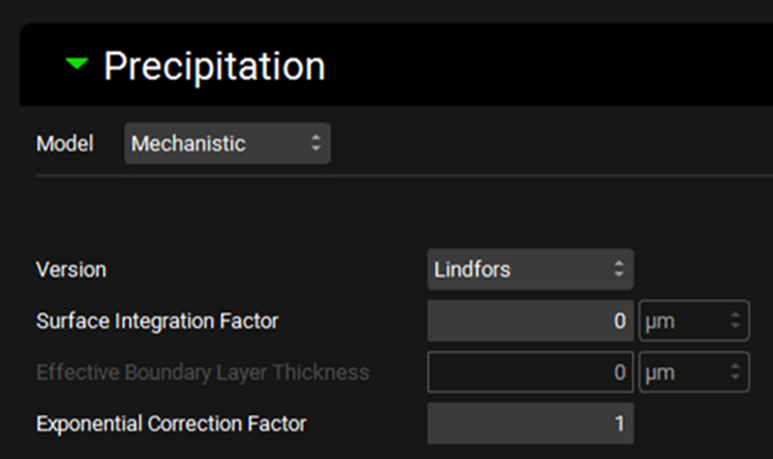
Simulations view, Compound Settings panel, Precipitation sub-panel (with Mechanistic model selected)
|
Input/Option |
Description |
|
Model |
A drop-down. Selects which precipitation model to use. First Order is the default selection. |
|
Version |
A drop-down. Selects which mechanistic model equations to use: Lindfors or SimulationsPlus. The default selection is Lindfors. |
|
Surface Integration Factor |
Defines the surface integration factor, in µm, for the Lindfors model. Higher values will result in a slower nucleation rate. |
|
Effective Boundary Layer Thickness |
Defines the Effective Boundary Layer Thickness, in µm, for the SimulationsPlus model. This term combines the surface integration factor and diffusion layer thickness terms. |
|
Exponential Correction Factor |
Defines the Exponential Correction factor for the Lindfors model. |
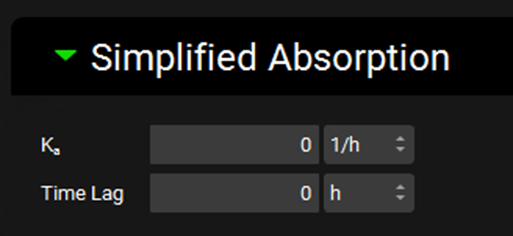
Simplified Absorption sub-panel
The Simplified Absorption sub-panel contains settings specific to simulations using the simplified absorption model. These parameters apply only to absorption, and do not relate to simplified systemic pharmacokinetic models. In the simplified absorption model, absorption refers to the movement of compound directly from the simplified dose compartment to the systemic circulation compartment (central compartment or venous return, depending on systemic model type).
If PKPlus™ (see PKPlus™ Module) is used to fit Ka and lag time to observed data, those parameters will be populated here upon export from PKPlus™.
Simulations view, Compound Settings panel, Simplified Absorption sub-panel
|
Input/Option |
Description |
|
Ka |
The absorption rate constant (Ka), defining the rate of absorption from the simplified dose compartment to systemic circulation. The value and the units (h-1, min-1, s-1) can be edited. |
|
Time Lag |
The lag time before absorption starts to occur. The value and the units (h, min, s) can be edited. |
Permeability sub-panel
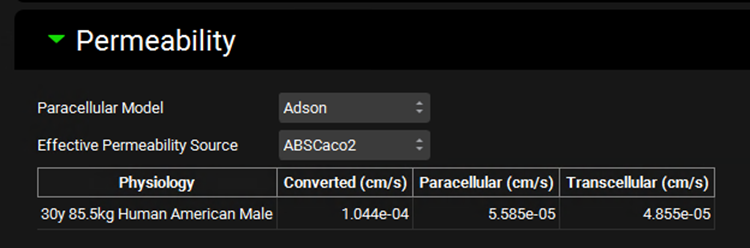
The Permeability sub-panel contains settings for paracellular and total effective jejunal permeability. The data input in Permeability panel on the Compounds View (see Permeability panel) is utilized here to calculate the converted total permeability for the indicated species. The breakdown between paracellular and transcellular permeability is then calculated based on the paracellular model selection and the gut properties of the physiology schedule selected for this simulation.
Simulations view, Compound Settings panel, Permeability sub-panel
|
Input/Option |
Description |
|
Paracellular Model |
A drop-down. Selects which paracellular model to use: Adson, Zhimin, or None. Adson is the default model for compounds added by the user. Zhimin is the default model for compounds imported through ADMET Predictor®. None turns off paracellular permeability for the simulation. |
|
Effective Permeability Source |
A drop-down. Selects the source, and by extension, the value, for the input permeability. The options available in this drop-down are based on the input permeabilities added to the table in the Permeability Panel on the Compounds view for the indicated compound (see Permeability panel). |
|
Physiology |
The first physiology in the Physiology Schedule selected for this simulation. It is provided for informational purposes, as the species of this physiology will dictate what interspecies conversion, if any, is applied to the permeability calculation. |
|
Converted |
The converted total effective jejunal permeability, in cm/s, that will be used in the simulation. |
|
Paracellular |
The paracellular component, in cm/s, of the converted total permeability. This is calculated based on the paracellular model indicated above. If the calculated value, based on either the Adson or Zhimin parameters of the compound, exceeds total permeability, this value will be capped at the total permeability value. |
|
Transcellular |
The transcellular component, in cm/s, of the converted total permeability. This is calculated as total permeability minus paracellular permeability. If total permeability minus calculated paracellular permeability would be less than 0, transcellular permeability is set to 0. |
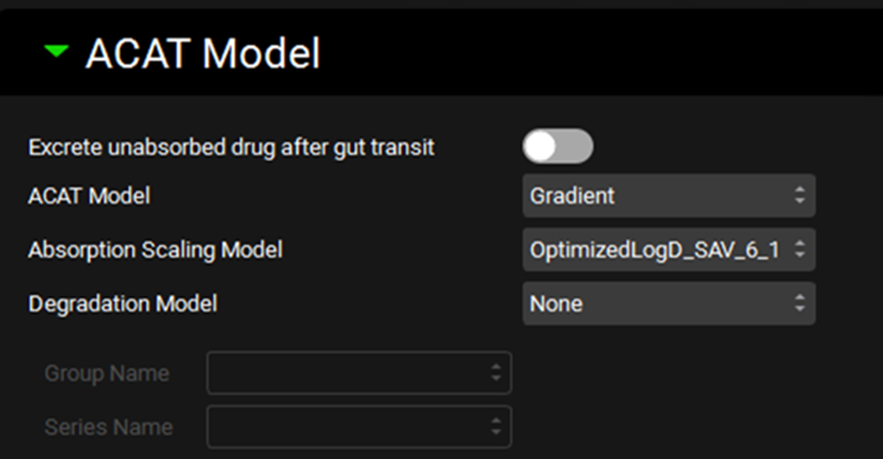
ACAT Model sub-panel
The ACAT Model sub-panel contains settings for the absorption and luminal degradation models in simulations using the ACAT Gastrointestinal Model (see Drug Administration Panel).
Simulations view, Compound Settings panel, ACAT Model sub-panel
|
Input/Option |
Description |
|
Excrete unabsorbed drug after gut transit |
A toggle, off by default. If toggled on, all drug will be immediately excreted at the end of the total gastrointestinal transit time. |
|
ACAT Model |
A drop-down. Selects the ACAT model type (Gradient or Unidirectional) to use for this simulation. Gradient is the default and the recommended model. In the Gradient model, drug transfer rates across apical and basolateral membranes of the enterocytes are calculated independently, and can go in either direction, depending on the concentration gradient. The Unidirectional model assumes that all drug that is absorbed moves instantly into the blood, permitting low-to-high absorption against a concentration gradient, and does not allow drug to be exsorbed. |
|
Absorption Scaling Model |
A drop-down. Selects which Absorption Scaling Factor model will be used to calculate the ASFs used in the simulation. Parameters for each ASF model can be set up and viewed on the Physiologies view in the Gastrointestinal panel. However, this setting controls which of those models is actually used in this simulation. |
|
Degradation Model |
A drop-down. Selects the model (Rate, Half-life, or None) to be used for chemical degradation in the gut lumen. The default setting in None. The Rate and Half-life models each require a corresponding Degradation vs Time series in Observed Data (see Profiles Panel), which is selected below. |
|
Group Name and Series Name |
Selects the Chemical Degradation Rate data from Observed Data to be used in either the Rate or the Half-life luminal degradation model. The Measurement Type must match the degradation model type. |
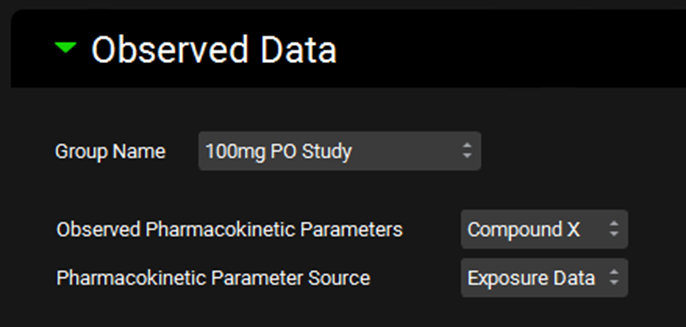
Observed Data sub-panel
The Observed Data sub-panel provides a means for linking observed pharmacokinetic data for a compound with the simulation. This linking is used to automatically display the corresponding observed data in certain key plots, to provide a source for observed summary pharmacokinetic parameters, and for mapping data for optimization. The identification of the type of data in an Exposure Data series (e.g. plasma concentration, amount in urine) must be done in the Observed Data view (see Observed Data View).
Simulations view, Compound Settings panel, Observed Data sub-panel
|
Input/Option |
Description |
|
Group Name |
A drop-down. Selects which group of Exposure Data will be associated with the indicated compound in this simulation. |
|
Observed Pharmacokinetic Parameters |
A drop-down. Selects which entry in the Parameters sub-panel (Observed Data view) will be associated with the indicated compound in this simulation. |
|
Pharmacokinetic Parameter Source |
A drop-down. Selects whether observed summary pharmacokinetic parameters will be derived from the Observed Pharmacokinetic Parameters (Parameters) entry or calculated from plasma concentration-time series within the linked Group (Exposure Data). Fa, Fdp, F, are always taken from the Parameters entry, if they exist. |
Additional Dosage Routes panel
The Additional Dosage Routes panel is used to view and set dosage route-specific simulation settings.
Please see:
and
within the Additional Dosage Routes Guide for details.
Configuration panel
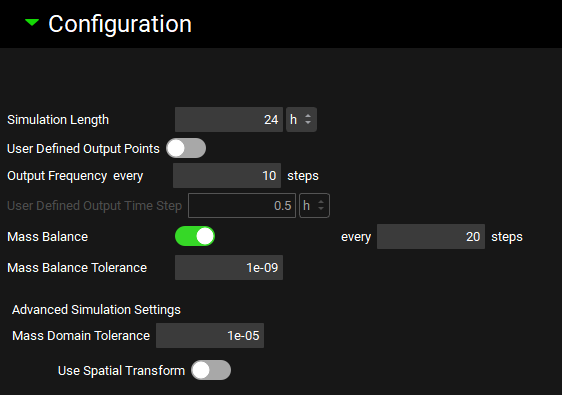
The Configuration panel is used to control simulation length, output settings, and integrator settings. The most common use of this panel is to set simulation length. The additional options are typically used either to modulate memory usage or to address individual issues with simulations experiencing integrator errors.
Simulations view, Configuration panel
|
Input/Option |
Description |
|
Simulation Length |
The time, in h, to which the simulation will be run. This value can be edited. |
|
User Defined Output Points |
A toggle. Off by default. When activated, allows the user to define the User Defined Output Time Step (see below). |
|
Output Frequency every [ ] steps |
The frequency of saved output data points in the simulated profile relative to the number of integrator time steps. With the default setting of 10, an output data point will be saved for every 10 integrator steps. This value can be edited. Decreasing this value will increase the number of output points and may result in increased memory consumption. |
|
User Defined Output Time Step |
The time step, in hours, between output points, as defined by the user. The value can be edited. Active only when the User Defined Output Points toggle is turned on. When this option is ON, the Output Frequency every [ ] steps will not be used to generate the simulated profiles. |
|
Mass Balance |
A toggle. Controls whether a mass balance check will be conducted during integration. |
|
Mass Balance every [ ] steps |
The number of integrator steps between mass balance checks. This value can be edited. Decreasing this value may increase the time it takes to run the simulation. |
|
Mass Balance Tolerance |
The cutoff, in g, above which a mass balance check is considered to have failed. This value can be edited. |
|
Advanced Simulation Settings |
Additional simulation settings that adjust the integrator settings. |
|
Mass Domain Tolerance |
How much mass, in g, is allowed to move into the negative domain in any one-time step during integration. Movement exceeding this tolerance will force the integrator to recalculate the step with a smaller step size. |
|
Use Spatial Transform |
A toggle. Controls whether negative mass values, within the domain tolerance, can be transformed to positive during integration in order to avoid integrator errors. Off by default. Turning on may help resolve certain integrator issues, but it may take simulations longer to run. |